Was verursacht Mukolipidose?
Mukolipidose (ML) ist eine seltene autosomal-rezessive lysosomale Speicherkrankheit. Rezessiv bedeutet, dass zwei Kopien des mutierten Gens (eine von jedem Elternteil) erforderlich sind, um die Störung zu verursachen. ML II und III stellen ein Spektrum von Schweregraden dar.
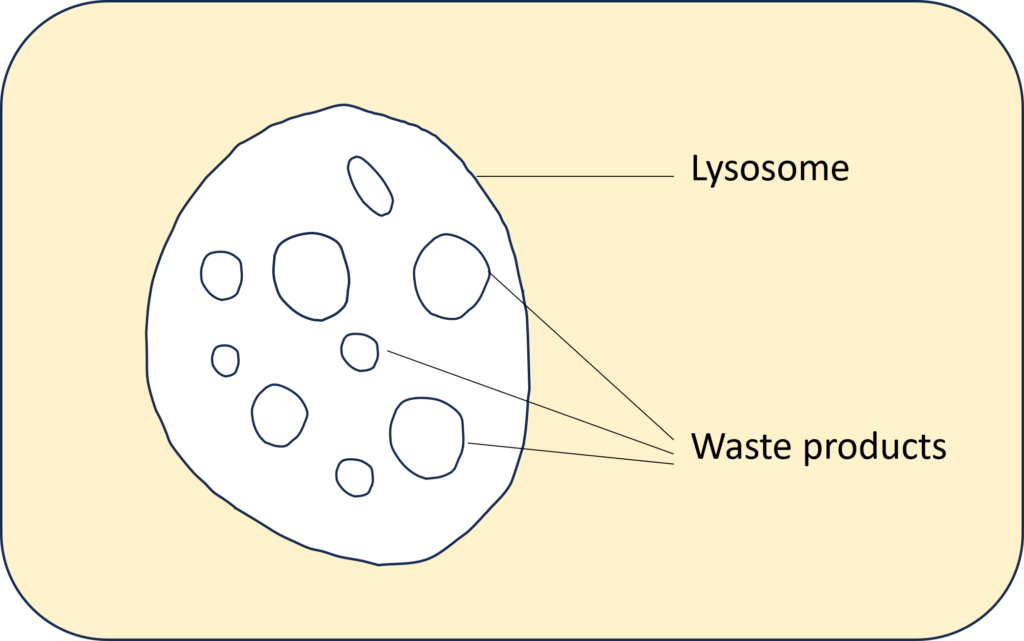
In jeder Zelle des Körpers gibt es Abfallprodukte, die beim Stoffwechsel anfallen. Diese Abfallprodukte werden in den Lysosomen gesammelt. Lysosomen fungieren als das Verdauungssystem der Zelle und dienen sowohl dem Abbau von Material, das von außerhalb der Zelle aufgenommen wurde, als auch der Verdauung veralteter Bestandteile der Zelle selbst.
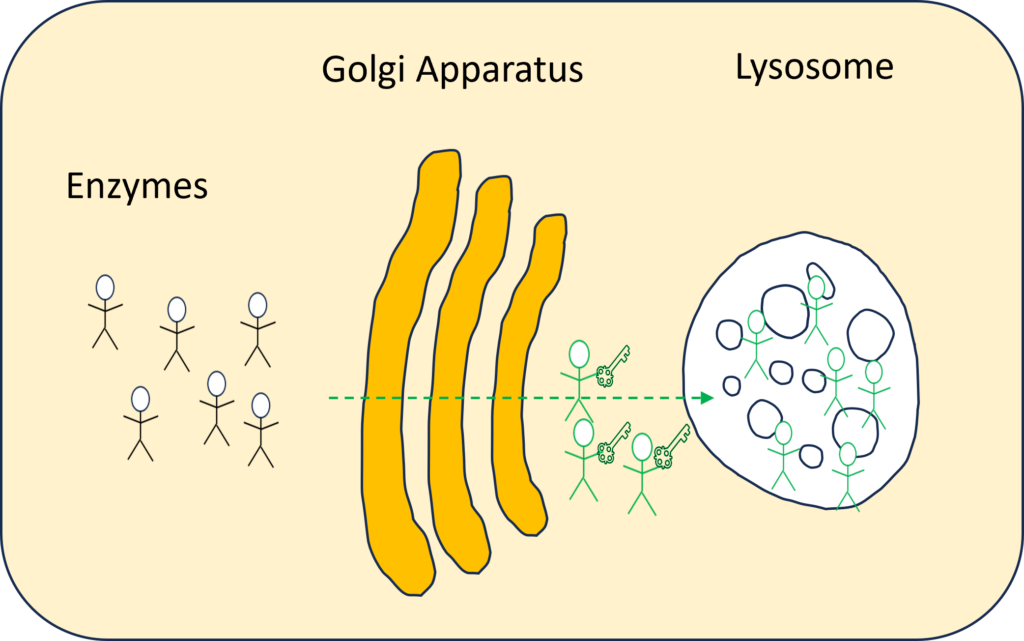
Innerhalb der Zelle gibt es eine Organelle, die Golgi-Apparat oder Golgi-Komplex genannt wird und als Fabrik fungiert, in der Proteine/Enzyme weiterverarbeitet und für den Transport zu ihren endgültigen Bestimmungsorten sortiert werden: Lysosomen, die Plasmamembran oder Sekretion. Es gibt 40 bis 60 Enzyme, die über den Golgi-Apparat zu den Lysosomen geschickt werden, um die Abfallprodukte zu recyceln. Im Golgi-Apparat erhält das Enzym, das gerade für das Recycling benötigt wird, den Schlüssel für das Lysosom.
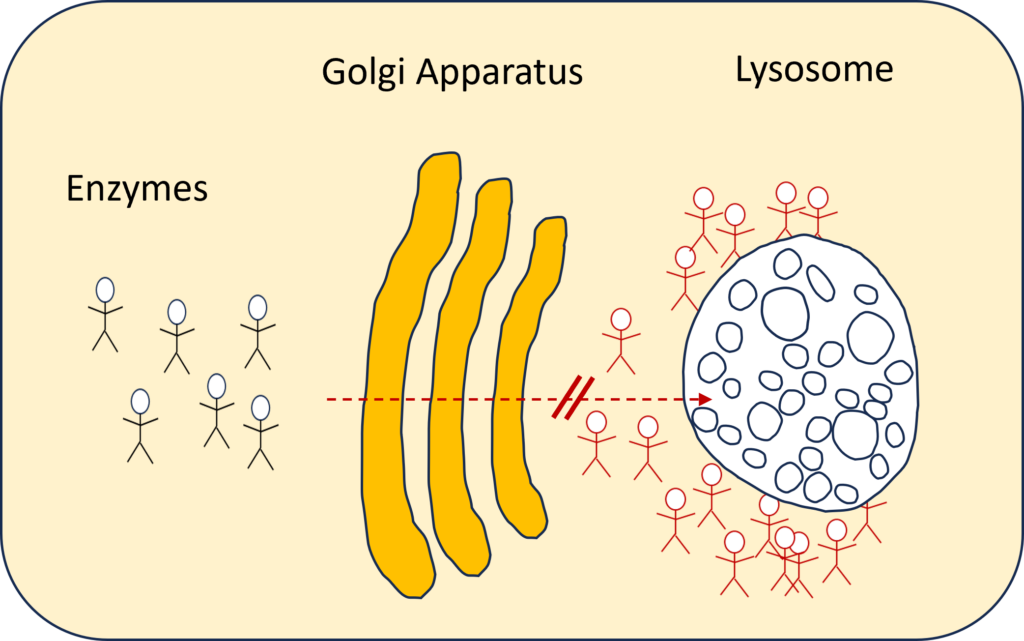
Im Falle von ML kann dieser Schlüssel, der für den Eintritt in die Lysosomen erforderlich ist, jedoch nicht an die Enzyme weitergegeben werden.
Technisch ausgedrückt: Die Mutationen im GNPTAB-Gen (ML-Typen II, II/III und III alpha/beta) führen zu einem Ausfall der Synthese von Mannose-6-Phosphat (MP6), dem Marker, der die Enzyme zu den Lysosomen führt. Ohne das MP6-Signal werden die Enzyme in den extrazellulären Raum fehlsortiert, was zu einer Anhäufung der Enzyme in den Geweben führt.
Daher schaffen es die lysosomalen Hydrolasen bei Menschen mit der Krankheit nicht bis zu den Lysosomen, was zu einer Insuffizienz der lysosomalen Enzyme führt. Ohne diese Enzyme sind die Zellen nicht in der Lage, bestimmte Substanzen wie Kohlenhydrate und Lipide abzubauen, was zu Anhäufungen führt, die Zell- und Gewebeschäden in mehreren Organen verursachen. [1]. Die Abfallprodukte stapeln sich im Lysosom, während sich die „arbeitslosen Enzyme” in und um die Zelle ansammeln. Aus diesem Grund wird sie auch als „Speicherkrankheit” bezeichnet.
Was sind die Symptome der Mukolipidose?
ML II
ML II (oder I-Zell-Krankheit) ist mit einem nahezu vollständigen Fehlen der Phosphotransferase-Aktivität verbunden, das auf Homozygotie oder Compound-Heterozygotie für Frameshift- oder Nonsense-Mutationen zurückzuführen ist.
Kinder, die mit Mukolipidose II geboren werden, sind bei der Geburt oft klein. Sie haben oft einen schwachen Schrei und einen geringen Muskeltonus (Hypotonie). Als Säuglinge wachsen sie langsam, bis ihre körperliche Entwicklung im Alter von etwa 24 Monaten zum Stillstand kommt.
Kinder mit Mukolipidose II können auch folgende Symptome aufweisen:
- verzögerte körperliche Entwicklung
- verzögerte kognitive Entwicklung
- verzögerte motorische Fähigkeiten
- Anomalien des Skeletts
- Abdominal- oder Nabelbruch
- Gelenkanomalien
- ausgeprägte, grobe Gesichtszüge
Weitere Symptome, die bei Kindern mit der Zeit auftreten können, sind:
- heisere Stimme aufgrund der Stimmbandversteifung
- häufige Infektionen der Atemwege
- häufige Ohrinfektionen, die zu Hörverlust führen
- übermäßiges Wachstum des Zahnfleischs (Gingivahyperplasie)
ML III a/β
Ursprünglich als PseudoHurler-Polydystrophie beschrieben. ML III behält eine geringe Phosphotransferase-Aktivität aufgrund von mindestens einer Missense- oder Spleißstellenmutation. Der Phänotyp ist milder, mit minimalen Verzögerungen in der Entwicklung, dem Auftreten einer Vergröberung des Gesichts im frühen Schulalter und einer Verlangsamung des Wachstums nach dem Alter von 4 Jahren [2].
ML II /III or Intermediate ML
Diese Gruppe von ML-Patienten weist einen ausgeprägten und konsistenten Phänotyp auf, der physische und röntgenologische Merkmale mit ML II teilt, aber in Bezug auf Lebenserwartung, phychomotorische Entwicklung und Grad der kognitiven Entwicklung eher dem Phänotyp ML III a/β ähnelt [3].
Prävalenz
Die Mukolipidose ist eine äußerst seltene Krankheit, und ihre genaue Prävalenz ist unbekannt.
Mukolipidose Typ II, Typ II/III und Typ III a/β & γ haben eine geschätzte kombinierte Inzidenz von 0,22 bis 2,70 pro 100.000 Lebendgeburten.
Es wird geschätzt, dass Mukolipidose Typ II weltweit bei etwa 1 von 100.000 bis 400.000 Personen auftritt.
Behandlung
Derzeit gibt es keine zugelassenen Therapien zur Umkehrung der Auswirkungen der Mukolipidose. Derzeitige Ansätze umfassen die Behandlung spezifischer Symptome durch gezielte Therapien und die Zusammenarbeit zwischen Spezialisten.
Referenzen
[1] https://www.tellmegen.com/en/results/monogenic-diseases/mucolipidosis-type-ii
[2] Cathey, S. S., Leroy, J. G., Wood, T., Eaves, K., Simensen, R. J., Kudo, M., … & Friez, M. J. (2010). Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. Journal of medical genetics, 47(1), 38-48.
[3] Leroy, J. G., Sillence, D., Wood, T., Barnes, J., Lebel, R. R., Friez, M. J., … & Cathey, S. S. (2014). A novel intermediate mucolipidosis II/IIIαβ caused by GNPTAB mutation in the cytosolic N-terminal domain. European Journal of Human Genetics, 22(5), 594-601.
Für weitere Informationen:
Dogterom, E. J., Wagenmakers, M. A., Wilke, M., Demirdas, S., Muschol, N. M., Pohl, S., … & Oussoren, E. (2021). Mucolipidosis type II and type III: a systematic review of 843 published cases. Genetics in Medicine, 23(11), 2047-2056.