What causes Mucolipidosis?
Mucolipidosis (ML) is a rare autosomal recessive lysosomal storage disorder. Recessive means that two copies of the mutated gene (one from each parent) are required to cause the disorder. ML II and III represent a spectrum of severity.
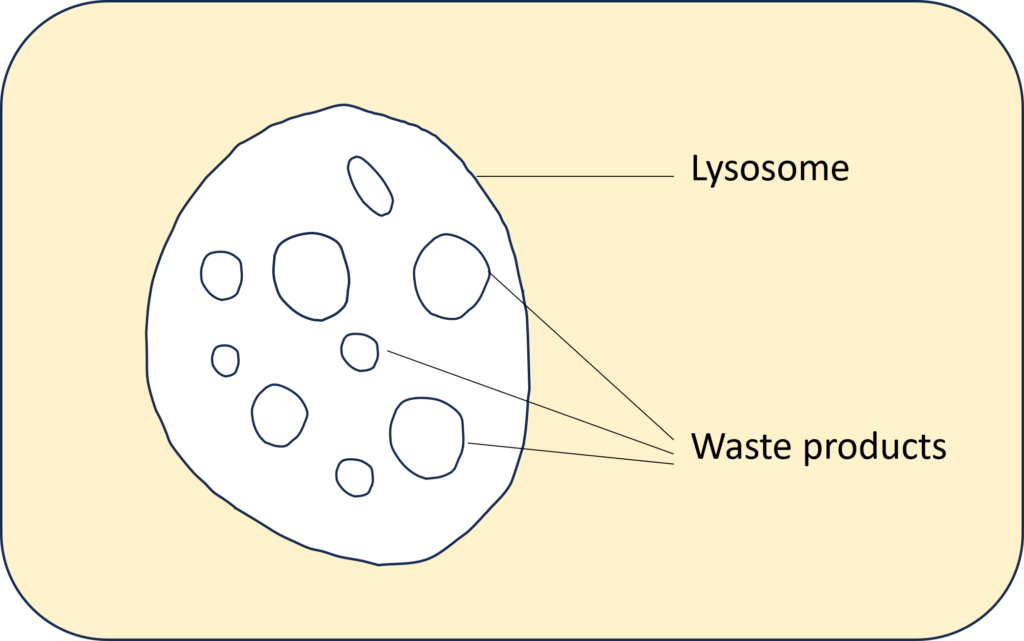
In every cell of the body there are waste products that arise from metabolism. These waste products are collected in the lysosome. Lysosomes function as the digestive system of the cell, serving both to degrade material taken up from outside the cell and to digest obsolete components of the cell itself.
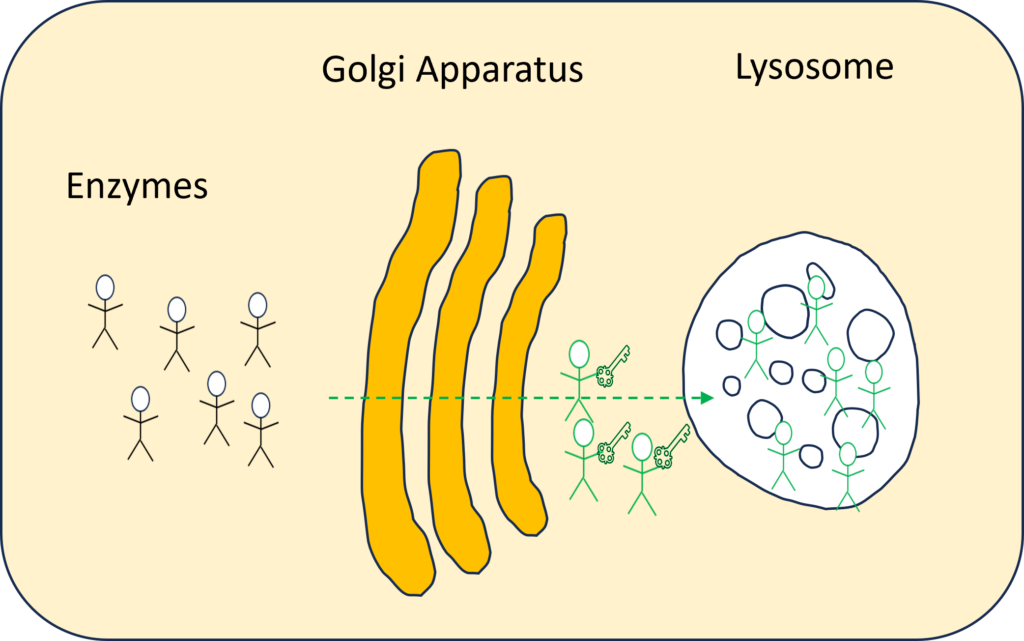
Within the cells there is an organelle called the Golgi apparatus, or Golgi complex, which functions as a factory in which proteins/ enzymes are further processed and sorted for transport to their eventual destinations: lysosomes, the plasma membrane, or secretion. There are 40 to 60 enzymes that are sent via the Golgi apparatus to the lysosome to recycle the waste products. In the Golgi apparatus, the enzyme that is currently needed for recycling receives the key for the lysosome.
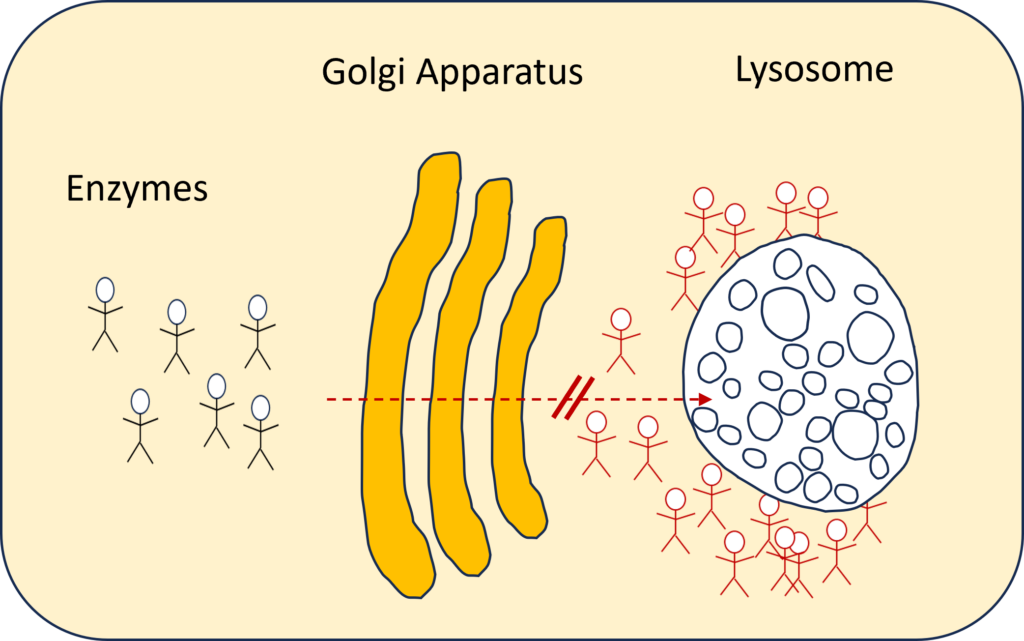
In case of ML, however, this key required to enter the lysosomes cannot be handed over to the enzymes.
In more technical terms: the mutations in the GNPTAB gene (ML types II, II/III and III alpha/beta) lead to a failure of mannose-6-phosphate (MP6) synthesis, the marker which targets the enzymes to the lysosomes. Without the MP6 signal, the enzymes are missorted into the extracellular space, leading to accumulation of the enzymes in the tissues.
Therefore, in individuals with the disease lysosomal hydrolases don’t make it to the lysosome resulting in multiple lysosomal enzyme insufficiency. Without the enzymes, “cells are unable to break down certain substances, such as carbohydrates and lipids, resulting in accumulations that induce cell and tissue damage in multiple organs”. [1]. The waste products pile up in the lysosome, while the “unemployed enzymes” accumulate in and around the cell. For this reason it is called a “storage disease”.
What are the symptoms of Mucolipidosis?
ML II
ML II ( or I-cell disease) correlates with near-total absence of phosphotransferase activity resulting from homozygosity or compound heterozygosity for frameshift or nonsense mutations.
Children born with mucolipidosis II are often small at birth. They often have a weak cry and poor muscle tone (hypotonia). As infants, they grow slowly until their physical development often plateaus around 24 months of age.
Children with mucolipidosis II may also have symptoms such as:
- delayed physical development
- delayed cognitive development
- delayed motor skills
- skeletal abnormalities
- abdominal or umbilical hernia
- joint abnormalities
- distinct facial features (coarse facial features)
Other symptoms children might experience over time include:
- hoarse voice due to vocal cord stiffening
- frequent respiratory infections
- frequent ear infections leading to hearing loss
- excessive growth of the gums (gingival hyperplasia)
ML III a/β
Originally described as PseudoHurler Polydystrophy. ML III retains a low level of phosphotransferase activity because of at least one missense or splice site mutation. The phenotype is milder, with minimal delays in milestones, the appearance of facial coarsening by early school age, and slowing of growth after the age of 4 years [2].
ML II /III or Intermediate ML
This group of ML patients has a distinct and consistent phenotype that shares physical and radiographic features with ML II, but more closely resembles the ML III a/β phenotype regarding life expectancy, phychomotor development and degree of cognitive development [3].
Prevalence
Mucolipidosis is an ultra rare disease and its exact prevalence is unknown.
Mucolipidosis type II, type II/ III and type III a/β & γ have an estimated combined incidence of 0.22 to 2.70 per 100,000 live births.
Mucolipidosis type II is estimated to occur in about 1 in 100,000 to 400,000 individuals worldwide.
Treatment
There are currently no approved therapies to reverse the effects of mucolipidosis. Current approaches involve managing specific symptoms through targeted therapies and collaboration between specialists.
References
[1] https://www.tellmegen.com/en/results/monogenic-diseases/mucolipidosis-type-ii
[2] Cathey, S. S., Leroy, J. G., Wood, T., Eaves, K., Simensen, R. J., Kudo, M., … & Friez, M. J. (2010). Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. Journal of medical genetics, 47(1), 38-48.
[3] Leroy, J. G., Sillence, D., Wood, T., Barnes, J., Lebel, R. R., Friez, M. J., … & Cathey, S. S. (2014). A novel intermediate mucolipidosis II/IIIαβ caused by GNPTAB mutation in the cytosolic N-terminal domain. European Journal of Human Genetics, 22(5), 594-601.
For further information:
Dogterom, E. J., Wagenmakers, M. A., Wilke, M., Demirdas, S., Muschol, N. M., Pohl, S., … & Oussoren, E. (2021). Mucolipidosis type II and type III: a systematic review of 843 published cases. Genetics in Medicine, 23(11), 2047-2056.